
M-CM (Macrocephaly-Capillary Malformation)
|
OMIM |
ORPHANET | CIE |
602501 |
600 |
Q87.3 |
Definición
Como su nombre indica, el síndrome Macrocefalia-Malformación capilar (M-MC) se distingue por macrocefalia (aumento del tamaño de la cabeza) y malformación capilar (manchas vasculares de nacimiento). Habitualmente se asocia con un crecimiento desproporcionado (“hemihiperplasia” o “hemihipertrofia”) de alguna parte del cuerpo.
La macrocefalia se debe a un crecimiento excesivo del cerebro, que además se puede asociar con anomalías de su desarrollo. El crecimiento excesivo del cerebro puede producir complicaciones como la hidrocefalia (retención y aumento del líquido cefalorraquídeo por dificultades en su circulación), o la compresión del tronco encefálico.
La malformación capilar da lugar a manchas vasculares de nacimiento, “reticuladas” o en forma de red, distribuidas por tronco y extremidades, que pueden cubrir una parte importante del cuerpo. Esta manchas tienden a atenuarse con el tiempo, lo que las diferencia de otras malformaciones vasculares como las “manchas de vino Oporto”. Es muy característica la presencia de una mancha vascular localizada entre el labio superior y la nariz.
Otras manifestaciones frecuentes de la M-MC incluyen la hiperlaxitud articular, una consistencia gomosa de la piel, y dedos supernumerarios (“polidactilia”) o fusionados entre sí (“sindactilia”).
La mayoría de los niños con M-CM presentan algún grado de retraso en el desarrollo psicomotor y discapacidad intelectual.
Diagnóstico
El diagnóstico de M-MC es fundamentalmente clínico y debe ser realizado por un especialista con experiencia y familiarizado con este tipo de trastornos. El diagnóstico se establece en base a las manifestaciones clínicas, aunque no es necesario que se presenten todas las características descritas. Además, el grado de afectación de cada persona puede ser muy variable.
PIK3CA, sobrecrecimiento y cáncer
El gen causante de M-CM, PIK3CA, que regula el crecimiento celular, se identificó en 2012. Las mutaciones de PIK3CA producen una mayor activación de su función que da lugar a un crecimiento excesivo de las células y tejidos afectados. Las mutaciones en PIK3CA se producen después de la fecundación, en las primeras fases de desarrollo del embrión. Surgen por primera vez en una célula que da lugar a una población de células hijas que se entremezclan con el resto de células normales, como las piezas de un mosaico; por eso se denomina “mosaicismo somático”. Esto explica la distribución parcheada y asimétrica de las manifestaciones clínicas, y el que la mutación en PIK3CA únicamente se detecte en el tejido afectado y no en la sangre circulante de una persona con M-MC.
PIK3CA es un gen está implicado en el desarrollo de tumores. A pesar de que por el momento no hay una evidencia definitiva de que M-MC se asocie con una mayor frecuencia de casos de cáncer, parece prudente mantener una cierta cautela hasta que se disponga de más información y recomendar el mismo protocolo de seguimiento para el diagnóstico precoz de tumor renal de Wilms que se emplea en otros trastornos genéticos con sobrecrecimiento, como el síndrome Beckwith-Wiedemann.
Clinica y Epidemiologia
El síndrome Macrocefalia-Malformación capilar es... Raro
Aproximadamente hay unos 150 casos documentados de M-CM en todo el mundo.
Poco conocido
M-CM fue descrito por primera vez en 1997. La gran mayoría de los casos diagnosticados hasta el momento han sido niños, de modo que la evolución de M-MC a largo plazo se desconoce; sobre todo cómo puede afectar a personas jóvenes y a adultos.
Genético pero no hereditario
M-CM es causado por una mutación en el gen PIK3CA que se produce después de la fecundación, en las primeras divisiones del cigoto, y afecta por tanto a una proporción variable de células del cuerpo; esto se conoce como “mosaicismo somático”. Por este motivo, la probabilidad que tienen los padres de un hijo con M-MC de tener otro hijo afectado en una siguiente gestación no es mayor que el del resto de la población general.
Variable
Las personas con M-CM pueden tener un grado de afectación muy variable entre ellos. El rango de discapacidad, tanto motora como cognitiva, puede oscilar entre leve y grave.
Compleja
La mayoría de las personas con M-MC requieren un seguimiento por diferentes especialistas (neurólogos y neurocirujanos, cirujanos generales y vasculares, traumatólogos y ortopedas, genetistas). Dependiendo de la presencia de otras anomalías asociadas puedes necesitar un seguimiento por otros especialistas (cardiólogos, endocrinólogos, ORL, oftalmólogos, etc.). Se recomienda específicamente realizar un seguimiento cercano de las posibles complicaciones del sobrecrecimiento cerebral (RM cerebral y de la médula/columna vertebral), y de tumor renal de Wilms (ecografías abdominales cada 4 meses hasta los 8 años.
Asesoramiento genético
El Síndrome de Macrocefalia-Malformación Capilar se presenta de forma esporádica por lo que no hay un riesgo aumentado de recurrencia en posteriores embarazos, ni en otros miembros de la familia.


Traducción: M-CM Network Syndrome Description
 |
Descripción del síndrome – Resumen General
La macrocefalia malformación capilar (M-CM) es un síndrome de malformación múltiple que causa sobrecrecimiento en cuerpo y cabeza y anomalías de la piel, el sistema vascular, el cerebro y las extremidades. El trastorno se ha atribuido recientemente (junio de 2012) a una mutación genética en un gen llamado PIK3CA. Se cree que la mutación en M-CM siempre ocurre después de que comienza la división celular y, por lo tanto, es muy poco probable que se herede. Una mutación que ocurre en esta etapa da como resultado diferentes porcentajes de células que se ven afectadas en diferentes individuos, por lo que existe una variabilidad significativa en la severidad con la que cada individuo se ve afectado.
Actualmente, el diagnóstico de M-CM se basa únicamente en observaciones clínicas. Aunque no todas las personas afectadas tienen todas las características, los signos comúnmente encontrados incluyen macrocefalia, macrosomia congénita, malformaciones capilares cutáneas extensas, asimetría corporal, dedos de manos o pies extra o fusionados, articulaciones laxas, piel pastosa, retrasos variables del desarrollo y otros problemas neurológicos como convulsiones y bajo tono muscular.
M-CM se describió por primera vez en la literatura médica en 1997. En ese momento se llamaba síndrome macrocefalia cutis marmorata telangiectásica congénita (M-CMTC) y este término se utiliza ocasionalmente. Recientemente se propuso un nuevo nombre, MCAP (2012), que se utiliza en algunos de los estudios de investigación sobre M-CM.
M-CM parece afectar a niños y niñas por igual y se ve entre todas las etnias. Ocurre esporádicamente en familias que no tienen otros familiares afectados. Si bien se han reportado menos de 300 casos de M-CM, es probable que haya muchas más personas afectadas que hayan sido diagnosticadas erróneamente, que no hayan sido reconocidas o que no hayan sido publicadas en la literatura médica.
El siguiente es un resumen detallado de M-CM que incluye una explicación de esta condición, hallazgos físicos, problemas médicos y opciones de tratamiento. Tenga en cuenta que no todos los niños con M-CM tendrán todos los problemas descritos. Al igual que cualquier persona con o sin una afección subyacente, cada paciente es diferente.
Cerebro
El crecimiento excesivo del cerebro y las anomalías son una característica principal y la causa de las complicaciones en M-CM. El tamaño anormalmente grande de la cabeza se llama macrocefalia. La principal causa de una gran cabeza en M-CM es la megalencefalia, aunque un problema adicional como la hidrocefalia puede hacer que la cabeza sea aún más grande.
Hidrocefalia.
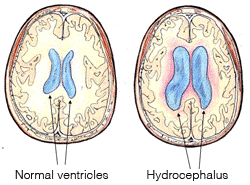
El cerebro humano es un órgano emparejado compuesto por dos mitades que se parecen. El líquido cefalorraquídeo (LCR) generalmente llena el espacio dentro de los ventrículos laterales.
La ventriculomegalia, o ventrículos laterales agrandados, se ve comúnmente en M-CM y es un término que a veces se usa indistintamente con hidrocefalia. La ventriculomegalia puede ser el resultado de la ampliación general del cerebro en M-CM. Cuando el agrandamiento se debe a una circulación deficiente del líquido cefalorraquídeo, y se acumula LCR causando una mayor presión en el cerebro, se presenta hidrocefalia.
A veces, un lado del cerebro es más grande que el otro y está mal formado, una afección llamada hemimegalencefalia. Si un lado es más grande pero no está mal formado en comparación con el lado más pequeño,
 esto se conoce como asimetría. Los términos hemimegalencefalia y asimetría cerebral a menudo se usan indistintamente, pero algunos sienten que esto no es preciso.
esto se conoce como asimetría. Los términos hemimegalencefalia y asimetría cerebral a menudo se usan indistintamente, pero algunos sienten que esto no es preciso.
Hernia amigdalar cerebelosa.
Otro hallazgo neurológico importante en M-CM es la hernia de las amígdalas cerebelosas (CTH, a menudo referida como Chiari). Al igual que con el resto del cerebro, el cerebelo tiende a crecer desproporcionadamente más rápido en niños con M-CM. La tasa de crecimiento del cerebro es demasiado rápida para que el cráneo se mantenga y se remodele. El cerebelo descansa en una cavidad en la parte posterior del cráneo llamada fosa posterior. La fosa posterior es un espacio restringido rodeado de hueso y un colgajo de tejido fibroso en la parte superior. A medida que el cerebelo crece, llena este espacio confinado. Cuando el cerebelo se agranda, las amígdalas cerebelosas sobresalen y se hernian a través del foramen magnum que conduce a CTH. Por lo tanto, el CTH generalmente se adquiere con el tiempo en M-CM a medida que crece el cerebro y es por eso que puede no estar presente en un recién nacido o en la primera infancia.
A veces, la CTH se describe como una malformación de Chiari I en estudios de imágenes cerebrales como la TC o la RM. Si bien las malformaciones tanto de CTH como de Chiari I implican herniación de las amígdala s cerebelosas a través del foramen magnum, no todos los casos de CTH son verdaderas malformaciones de Chiari I. Clásicamente, una malformación de Chiari I se describe en imágenes cerebrales como una hernia de tonsilas cerebelares de más de 5 milímetros por debajo del foramen magnum. Esta definición, sin embargo, no aclara la causa subyacente. Una malformación de Chiari I es causada por una fosa posterior pequeña o anormalmente formada y, por lo tanto, es un defecto típicamente presente desde el nacimiento. Una malformación congénita de Chiari I puede ser un hallazgo aislado en un individuo por lo demás normal, pero también puede asociarse con otras afecciones. Una malformación aislada de Chiari I puede no mostrar signos o síntomas y, a menudo, se descubre incidentalmente cuando se realiza una tomografía computarizada o una resonancia magnética por alguna otra razón.
s cerebelosas a través del foramen magnum, no todos los casos de CTH son verdaderas malformaciones de Chiari I. Clásicamente, una malformación de Chiari I se describe en imágenes cerebrales como una hernia de tonsilas cerebelares de más de 5 milímetros por debajo del foramen magnum. Esta definición, sin embargo, no aclara la causa subyacente. Una malformación de Chiari I es causada por una fosa posterior pequeña o anormalmente formada y, por lo tanto, es un defecto típicamente presente desde el nacimiento. Una malformación congénita de Chiari I puede ser un hallazgo aislado en un individuo por lo demás normal, pero también puede asociarse con otras afecciones. Una malformación aislada de Chiari I puede no mostrar signos o síntomas y, a menudo, se descubre incidentalmente cuando se realiza una tomografía computarizada o una resonancia magnética por alguna otra razón.
La hernia tonsilar cerebelosa, independientemente de la causa subyacente, puede provocar compresión o presión sobre el tallo cerebral. La hernia también puede causar un flujo reducido de LCR a través del foramen magnum, lo que lleva a un aumento de la presión dentro del cerebro. No todos tendrán síntomas, pero los signos importantes de la compresión del tronco encefálico incluyen:
- convulsiones
- apnea
- dificultad para controlar los movimientos oculares
- letargo
- irritabilidad
- dolores de cabeza
- dolor de cuello
- problemas de equilibrio
- debilidad muscular
- entumecimiento
- hormigueo en brazos o piernas
- mareo
- problemas de la vista
- dificultad para tragar
- zumbido o zumbido en los oídos
- pérdida de la audición
- vómitos
- insomnio
- cambios en las habilidades motoras finas
- depresión
Los bebés pueden tener dificultad para tragar, irritabilidad cuando se les alimenta, babeo excesivo, un llanto débil, náuseas o vómitos, debilidad en el brazo, rigidez en el cuello, problemas para respirar y retrasos en el desarrollo. Si un niño tiene alguno de estos u otros síntomas preocupantes, es importante buscar atención médica de inmediato. Si los signos no se reconocen temprano y se tratan de inmediato, los niños pueden sufrir complicaciones graves, incluida la muerte súbita.
Otros hallazgos neurológicos comunes en M-CM incluyen:
- displasia cortical focal
- polimicrogiria Este hallazgo generalmente se asocia con el funcionamiento anormal del cerebro y puede coexistir con retraso mental y convulsiones.
- anomalías de la sustancia blanca que pueden representar mielinización retardada o anormal. A veces, estos cambios pueden parecerse a una leucodistrofia. Si bien estos hallazgos no necesariamente causan problemas funcionales en M-CM, tales cambios en la sustancia blanca pueden sugerir un proceso de maduración cerebral anormal o incluso deterioro del cerebro en otros trastornos.
- cuerpo calloso grueso En M-CM, esta estructura parece más grande de lo normal. Se desconoce si este hallazgo contribuye al funcionamiento neurológico anormal.
 La mayoría de los niños con M-CM tienen hipotonía. La hipotonía puede ser causada por problemas en el cerebro o el músculo y se ve como parte de muchas condiciones diferentes que afectan el control muscular. Se cree que la hipotonía en M-CM se debe a problemas dentro del cerebro y es otro factor que contribuye a la demora en el desarrollo de los niños afectados. La hipotonía en M-CM a menudo mejora con la edad, sin embargo, los problemas neurológicos subyacentes pueden causar que esto sea un problema continuo incluso en niños mayores.
La mayoría de los niños con M-CM tienen hipotonía. La hipotonía puede ser causada por problemas en el cerebro o el músculo y se ve como parte de muchas condiciones diferentes que afectan el control muscular. Se cree que la hipotonía en M-CM se debe a problemas dentro del cerebro y es otro factor que contribuye a la demora en el desarrollo de los niños afectados. La hipotonía en M-CM a menudo mejora con la edad, sin embargo, los problemas neurológicos subyacentes pueden causar que esto sea un problema continuo incluso en niños mayores.
Manejo neurológico y tratamiento
Las directrices de gestión recientemente publicadas requieren una RM cerebral y de columna vertebral a l momento del diagnóstico, luego cada 6 meses de 0 a 2 años y cada año a partir de 2 a 6 años con imágenes más frecuentes si surgen problemas particulares. A los seis años, el rápido aumento en la circunferencia de la cabeza se ha ralentizado y las preocupaciones con el crecimiento del cerebelo disminuyen. Las imágenes más allá de los 6 años de edad deben depender de los hallazgos previos y el curso clínico. Aunque la hidrocefalia se ve comúnmente en M-CM, la colocación de una derivación ventriculperitoneal (VP) no corrige completamente la macrocefalia en individuos afectados porque la causa principal de la macrocefalia en M-CM es el crecimiento excesivo del cerebro. Puede reducir el grado de macrocefalia y con el tiempo puede disminuir la velocidad con la que la cabeza se agranda en algunas personas afectadas, pero no en todas. Algunos también han tratado la hidrocefalia con éxito con una tercera ventriculostomía endoscópica (ETV). Este procedimiento elimina la necesidad de colocar objetos extraños, como los tubos necesarios para la derivación VP, lo que teóricamente reduce las complicaciones después de la cirugía. Sin embargo, no existe un consenso claro sobre el mejor tratamiento quirúrgico para la hidrocefalia en M-CM. Los pacientes con M-CM con derivación de ETV y VP han requerido revisiones.
l momento del diagnóstico, luego cada 6 meses de 0 a 2 años y cada año a partir de 2 a 6 años con imágenes más frecuentes si surgen problemas particulares. A los seis años, el rápido aumento en la circunferencia de la cabeza se ha ralentizado y las preocupaciones con el crecimiento del cerebelo disminuyen. Las imágenes más allá de los 6 años de edad deben depender de los hallazgos previos y el curso clínico. Aunque la hidrocefalia se ve comúnmente en M-CM, la colocación de una derivación ventriculperitoneal (VP) no corrige completamente la macrocefalia en individuos afectados porque la causa principal de la macrocefalia en M-CM es el crecimiento excesivo del cerebro. Puede reducir el grado de macrocefalia y con el tiempo puede disminuir la velocidad con la que la cabeza se agranda en algunas personas afectadas, pero no en todas. Algunos también han tratado la hidrocefalia con éxito con una tercera ventriculostomía endoscópica (ETV). Este procedimiento elimina la necesidad de colocar objetos extraños, como los tubos necesarios para la derivación VP, lo que teóricamente reduce las complicaciones después de la cirugía. Sin embargo, no existe un consenso claro sobre el mejor tratamiento quirúrgico para la hidrocefalia en M-CM. Los pacientes con M-CM con derivación de ETV y VP han requerido revisiones.
Cuando el sobrecrecimiento excesivo del cerebro y el CTH causan síntomas de compresión del tallo encefálico, algunos pacientes con M-CM han sido tratados con éxito con un procedimiento llamado descompresión de la fosa posterior. Este procedimiento agranda quirúrgicamente la abertura en la base del cráneo, permitiendo que el cerebelo crezca en un espacio que ahora puede acomodar su tamaño. El objetivo de este 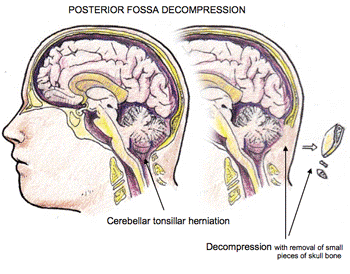 procedimiento es resolver o prevenir los síntomas debido a la compresión del tronco encefálico. La presencia de CTH aumenta el riesgo de siringomielia, una afección en la que se forma un quiste, denominado siringe, en la médula espinal. Se puede detectar una siringe a través de una RM espinal. Esta condición requiere control y, en ocasiones, tratamiento quirúrgico. Muchas personas que tienen CTH, un cerebelo demasiado grande y ventrículos agrandados requerirán neurocirugía en algún momento. El momento y el tipo de intervención es un asunto delicado y se determina mejor caso por caso. Las opciones de neurocirugía dependen de una serie de factores que incluyen la gravedad de las anomalías cerebrales, las opiniones de los médicos directamente involucrados en la atención de esa persona y los síntomas.
procedimiento es resolver o prevenir los síntomas debido a la compresión del tronco encefálico. La presencia de CTH aumenta el riesgo de siringomielia, una afección en la que se forma un quiste, denominado siringe, en la médula espinal. Se puede detectar una siringe a través de una RM espinal. Esta condición requiere control y, en ocasiones, tratamiento quirúrgico. Muchas personas que tienen CTH, un cerebelo demasiado grande y ventrículos agrandados requerirán neurocirugía en algún momento. El momento y el tipo de intervención es un asunto delicado y se determina mejor caso por caso. Las opciones de neurocirugía dependen de una serie de factores que incluyen la gravedad de las anomalías cerebrales, las opiniones de los médicos directamente involucrados en la atención de esa persona y los síntomas.
Discapacidades de aprendizaje, retrasos y deterioro cognitivo
Además de las anomalías cerebrales estructurales, se observan problemas funcionales en casi todos los individuos con M-CM. Las discapacidades del desarrollo son comunes y pueden variar desde una discapacidad leve, moderada a ocasionalmente grave. Parece que los niños con trastornos de formación cerebral como polimicrogiria y displasia cortical tienen un deterioro más grave; sin embargo, no se han realizado estudios a largo plazo para aclarar esta asociación.
Los retrasos psicomotrices son comunes e influenciados por múltiples factores, incluyendo articulaciones sueltas, asimetría de la extremidad y un tamaño grande de cabeza y cuerpo. En casos típicos, los niños aprenden a caminar y hablar, pero a una edad posterior a la esperada. La mayoría de los niños requieren terapias especiales que incluyen físioterapia, ocupacional y del habla. Es importante que las personas con M-CM y sus familias trabajen estrechamente con los recursos en su área para garantizar que se reciban los servicios apropiados.
Se prevé que los niños con M-CM tendrán discapacidades de por vida. Sin embargo, es importante recordar que, en última instancia, cada niño tendrá su propio destino de desarrollo. Los niños con M-CM deben tener todas las oportunidades para sobresalir dentro de sus propias limitaciones. Debido a que se han reportado tan pocos casos de adultos, no se puede predecir con exactitud qué tan retrasados o funcionales serán los niños con M-CM a medida que maduren.
En general, los niños con M-CM son sociales, felices y juguetones. Disfrutan ser parte de sus familias y tener interacciones significativas con los padres, la familia y los amigos.
Con base en la información disponible, M-CM no parece ser una condición asociada con la regresión o disminución en el funcionamiento mental de una persona. Se debe esperar que los niños progresen lenta y constantemente en el desarrollo; sin embargo, también se debe esperar que estén detrás de sus pares hasta cierto punto. El autismo no es una característica típica, pero se ha observado en algunos casos.
Riesgos de cáncer
Los niños con el síndrome de Beckwith-Wiedemann, otro síndrome de sobrecrecimiento asociado con la hemihiperplasia, tienen un mayor riesgo de ciertos tipos de cáncer de hígado (hepatoblastoma) y cáncer de riñón (tumor de Wilms). Si bien M-CM es una condición de sobrecrecimiento, actualmente no está claro si existe realmente un mayor riesgo de estos cánceres. En informes publicados, ha habido dos casos de individuos con M-CM que luego se descubrió que tenían un tumor de Wilms. No se han reportado casos de hepatoblastoma.
La mutación genética que ahora se piensa que causa M-CM, PIK3CA, se encuentra mutada en el cáncer. Además, PIK3CA se ha implicado recientemente en el síndrome CLOVES, donde se ha demostrado con mayor claridad el riesgo de un tumor de Wilms.
Recientemente, los investigadores hicieron la recomendación de detectar el tumor de Wilms siguiendo las pautas del síndrome de Beckwith-Wiedemann: ultrasonidos renales cada 3 meses hasta los 8 años. La prueba de sangre AFP indicada para el síndrome de Beckwith-Wiedemann para detectar hepatoblastoma (cáncer de hígado) no se recomienda para pacientes con M-CM en este momento.
La literatura médica para M-CM informa un caso de leucemia y dos de meningiomas (tumores cerebrales).
Crecimiento
El crecimiento excesivo es una característica importante de M-CM. Además del sobrecrecimiento cerebral, se manifiesta como macrosomía prenatal y hemihiperplasia.
La macrosomía prenatal es muy común, pero la mayoría de los niños con M-CM vuelven a caer dentro del rango normal durante los primeros dos años y continúan creciendo a una velocidad normal a partir de entonces.
A veces el crecimiento de los niños cae por debajo de las curvas normales. Cuando esto se deba a problemas de alimentación, se pueden recomendar dietas modificadas o el uso de un tubo gástrico. Algunos niños muestran signos de problemas endocrinos, como hipoglucemia o retraso en la edad ósea. Estos niños deben ser evaluados por un endocrinólogo pediátrico. Se ha encontrado que algunos niños con M-CM tienen una deficiencia de la hormona del crecimiento.
Hasta la fecha, no se han realizado estudios clínicos que analicen los riesgos y beneficios de la terapia con hormona de crecimiento en pacientes con M-CM, ni hay informes de casos que describan los efectos y resultados de la terapia con hormona de crecimiento en pacientes con M-CM. No todos los pacientes con M-CM y una deficiencia documentada de la hormona de crecimiento se les ha recetado la hormona del crecimiento. Los proveedores deben tener en cuenta al considerar la hormona del crecimiento que puede haber un aumento de la predisposición tumoral en el síndrome.
Hemihiperplasia o asimetría es una característica común y una pista importante para un diagnóstico de M-CM. El grado de asimetría varía entre las personas afectadas y, por lo general, permanece proporcionado a lo largo de la vida. Esto significa que la extremidad más grande seguirá siendo más grande, pero no continuará creciendo más rápido que el resto del cuerpo a medida que los niños con M-CM envejecen.
La hemihiperplasia no es específica de M-CM; puede verse en otros síndromes de sobrecrecimiento o como una ocurrencia aislada.
Piel y vascular
Resultados de la piel en M-CM
 En M-CM, se encuentran comúnmente dos tipos de marcas de nacimiento vasculares. Una es una decoloración difusa, vívida y de color rojo violáceo con márgenes mal definidos en una distribución irregular o desigual en todo el cuerpo. Esto se presenta en los recién nacidos afectados y muy a menudo se parece a un tipo vivo de cutis marmorata. Este hallazgo de piel muy distintivo se conoce como una malformación capilar. Como hallazgos aislados, las malformaciones capilares son muy comunes en los recién nacidos normales. Los términos laicos usados para tales lesiones faciales incluyen "mordida de cigüeña" cuando ocurre en la nuca o "besos de ángel" cuando se ven en la frente o el párpado. Pueden ser referidos por los médicos o en la literatura médica como "nevus flammeus" o "angiomata". Estos hallazgos pueden oscurecerse con los cambios de llanto o posición.
En M-CM, se encuentran comúnmente dos tipos de marcas de nacimiento vasculares. Una es una decoloración difusa, vívida y de color rojo violáceo con márgenes mal definidos en una distribución irregular o desigual en todo el cuerpo. Esto se presenta en los recién nacidos afectados y muy a menudo se parece a un tipo vivo de cutis marmorata. Este hallazgo de piel muy distintivo se conoce como una malformación capilar. Como hallazgos aislados, las malformaciones capilares son muy comunes en los recién nacidos normales. Los términos laicos usados para tales lesiones faciales incluyen "mordida de cigüeña" cuando ocurre en la nuca o "besos de ángel" cuando se ven en la frente o el párpado. Pueden ser referidos por los médicos o en la literatura médica como "nevus flammeus" o "angiomata". Estos hallazgos pueden oscurecerse con los cambios de llanto o posición.
En los niños con M-CM, este tipo de malformación capilar a menudo es más dramático y pronunciado. A medida que el niño crece, la región afectada se atenúa a un rojo rosado, y con el tiempo las malformaciones pueden ser aún menos evidentes o notarse solo cuando el niño está frío o tiene fiebre. A veces, un lado o parte del cuerpo puede verse más afectado por estas malformaciones capilares que otro. En algunos niños, hay una demarcación clara y abrupta de malformaciones capilares en la línea media del cuerpo. Observaciones como esta han llevado a algunos a concluir que esto es evidencia de que el cambio genético que causa M-CM es mosaico (lo que significa que el cambio genético existe en algunas células del cuerpo, pero no en otras).
Las malformaciones capilares en M-CM a veces se refieren incorrectamente como una mancha de vino de Oporto. Si bien una mancha de vino de Oporto es un tipo de malformación capilar, se vuelve más oscura con el tiempo, mientras que las malformaciones capilares observadas en M-CM se desvanecen con el tiempo. Este hallazgo cutáneo en individuos con M-CM puede ser una manifestación fíisica alarmante del síndrome, pero de hecho no suele causar ninguna molestia o problemas médicos. Se han informado algunos niños con un diagnóstico clínico de M-CM que no han observado malformaciones capilares, lo que sugiere que el aspecto vascular de la piel puede desaparecer por completo con el tiempo en algunas personas. No se sabe si los casos informados tuvieron algún cambio vascular o si fue solo un hallazgo sutil al nacer que se desvaneció con el tiempo.
El otro hallazgo notable en la piel del recién nacido en M-CM incluye una marca de nacimiento de color rojo oscuro que se ve frecuentemente en la línea media de la cara, el surco y / o el labio superior. Esta marca de nacimiento se considera una malformación capilar en la línea media. Se puede confundir con una mancha de vino de Oporto o incluso con un hemangioma (un tipo de tumor vascular), pero estos términos serían incorrectos ya que permanece plano y se desvanece con el tiempo. Cuando se observan, estas malformaciones capilares en la línea media con frecuencia llevan a los médicos a considerar el diagnóstico de M-CM. Otras áreas de la cara que pueden tener estas anomalías en M-CM incluyen el labio inferior, los párpados y la frente. En su mayor parte, estas malformaciones capilares faciales parecen ser solo un problema cosmético a pesar de su aspecto alarmante y no representan riesgos adicionales para la salud. Si bien se puede considerar la terapia con láser para ayudar a reducir su visibilidad, el alcance y la ubicación de las lesiones junto con el hecho de que generalmente se desvanecen con el tiempo son factores importantes que los padres deben considerar al considerar este tratamiento.
Al principio de su descubrimiento, se consideró que los cambios en la piel de M-CM representaban o se asemejaban mucho a una condición llamada CMTC (cutis marmorata telangiectásica congénita) y la información sobre CMTC se encuentra a menudo en las búsquedas en Internet para M-CM. En 2007, el nombre se cambió de M-CMTC a M-CM para reflejar con mayor precisión las anomalías vasculares del trastorno. CMTC es una rara enfermedad que afecta principalmente a los vasos sanguíneos de la piel. Por lo general, se ve al nacer o poco después y causa un veteado de color púrpura de la piel que se parece al cutis marmorata, pero es mucho más pronunciado. Este marmoleado puede limitarse solo a una determinada parte del cuerpo o puede distribuirse en áreas extensas. Mientras que las personas con CMTC pueden tener retrasos en el desarrollo, no tienen los problemas neurológicos únicos o anomalías del desarrollo cerebral que se observan en M-CM. Además, mientras que M-CM puede tener un sobrecrecimiento asociado de partes del cuerpo afectadas (hemihiperplasia), CMTC más típicamente involucra sotobosque (hipoplasia) de las extremidades o partes del cuerpo afectadas por la anomalía vascular. Mientras que las anomalías vasculares vívidas y difusas de M-CM pueden parecerse a CMTC, estas condiciones parecen ser entidades separadas y distintas que tienen diferencias obvias y no hay evidencia de que exista una asociación genética o clínica entre las dos afecciones más allá de la piel.En resumen, los hallazgos vasculares y cutáneos en M-CM son complejos y aún se comprenden muy poco. Una descripción precisa de los hallazgos cutáneos de un individuo y las anomalías vasculares es importante para proporcionar pistas diagnósticas adecuadas que reduzcan el riesgo de un diagnóstico erróneo. Consulte más abajo sobre los hallazgos vasculares en M-CM.
Antecedentes y nomenclatura
Las malformaciones vasculares a menudo son reconocidas por los padres o médicos como una especie de marca de nacimiento. Con los años, los términos utilizados para describir ciertas malformaciones vasculares en individuos con y sin M-CM han sido inconsistentes u ocasionalmente confusos. Se ha creado un mejor sistema de clasificación para definir con mayor precisión estos hallazgos cutáneos.
Respecto a las anomalías vasculares comunes de la piel, existen dos tipos de anormalidades que se encuentran comúnmente en los bebés: los tumores vasculares (como hemangiomas) y las malformaciones vasculares. Los hemangiomas son crecimientos benignos de vasos sanguíneos. Pueden o no estar presentes en el momento del nacimiento, por lo general crecen en la infancia, y luego a menudo regresan. Si bien este tipo de anomalía vascular se puede ver en M-CM, no son necesariamente características de la condición y muchos niños con M-CM no tienen hemangiomas verdaderos. Algunos datos sugieren que puede haber una tendencia a que estos crecimientos vasculares ocurran dentro del cuerpo u otros tejidos de niños con M-CM a medida que envejecen, pero la historia natural de estos hemangiomas internos no está bien definida y su frecuencia y consecuencias en M-CM es poco conocido.
A diferencia de los hemangiomas, las malformaciones vasculares son básicamente colecciones de vasos sanguíneos anormalmente formados que existen como defectos localizados cerca de la superficie de la piel. A diferencia de los hemangiomas, las malformaciones vasculares siempre son congénitas y están presentes en el momento del nacimiento, aunque es posible que no se reconozcan inmediatamente en la inspección debido a la variabilidad en su apariencia. Estas lesiones no crecen ni retroceden, pero ciertos tipos de estas malformaciones pueden desvanecerse con el tiempo. Ocasionalmente, tales malformaciones vasculares pueden ser referidas incorrectamente como "hemangiomas" incluso por los proveedores de atención médica, lo que a veces genera confusión cuando se describen estas anomalías.
Las malformaciones vasculares se subdividen en lesiones de flujo rápido y flujo lento dependiendo de la velocidad a la que la sangre fluye a través de ellas. Las malformaciones capilares son un tipo de malformación vascular de flujo lento que afecta la formación de los capilares. En general, las malformaciones capilares se pueden categorizar por su ubicación en la cara.
Las malformaciones capilares medias (línea media) se encuentran a lo largo del centro o en el medio de la cara. Por lo general, tienen la apariencia de un parche de color rosa salmón (o salmón) plano. Como hallazgos aislados, estos son muy comunes en niños recién nacidos normales. Los términos laicos usados para tales lesiones faciales incluyen "mordida de cigüeña" cuando ocurre en la nuca o "besos de ángel" cuando se ven en la frente o el párpado. Los médicos o en la literatura médica pueden referirlos como "nevus flammeus" "O" angiomata ". La historia natural de estos "parches de salmón"es que a menudo se desvanecen con el tiempo (particularmente en los primeros años de vida), o al menos se vuelven menos notorios.
Las malformaciones capilares laterales pueden ocurrir a ambos lados de la cara, pero no suelen encontrarse a lo largo de la línea media. Estas lesiones son a menudo de color más oscuro que las manchas de salmón y se consideran "manchas de vino de Oporto". Las manchas de vino de Oporto son marcas de nacimiento planas llenas de pequeños vasos sanguíneos cerca de la superficie de la piel causando una decoloración rojiza o violácea. Por lo general, son de color rosa en los primeros años de la vida y adquieren un color más intenso a medida que el niño madura. No se desvanecen o desaparecen y el área de la piel afectada crece en proporción al resto del cuerpo a medida que el niño crece. En M-CM, este tipo específico de malformación capilar no es tan común como las malformaciones capilares de la línea media; sin embargo, una persona con M-CM puede tener varios tipos de malformaciones vasculares que ocurren en cualquier parte del cuerpo.
Vascular
Las malformaciones vasculares anormales que se manifiestan en la piel de las personas con M-CM también pueden estar presentes en las profundidades del cuerpo. La evidencia sugiere que existe un mayor riesgo de malformaciones de grandes vasos, como las arterias o venas que llevan sangre al corazón y lejos del mismo. Esto puede correlacionarse con el mayor riesgo de defectos cardíacos congénitos en este síndrome, ya que el desarrollo de estas estructuras en la vida fetal está relacionado.
Según los informes de casos, parece haber un mayor riesgo de accidentes cerebrovasculares o eventos tromboembólicos en individuos con M-CM, aunque esto parece ser en la minoría de los casos. La literatura sugiere que estos eventos trombóticos pueden afectar los vasos sanguíneos, ya sea en el cerebro o en otras partes del cuerpo. La razón exacta del aumento del riesgo de accidentes cerebrovasculares en M-CM no está clara; se puede relacionar con los vasos sanguíneos anormalmente formados. Debido a que los accidentes cerebrovasculares pueden causar síntomas graves o incluso potencialmente mortales, es importante tratar cualquier síntoma de preocupación de inmediato. Los signos y síntomas de un accidente cerebrovascular que afectan el cerebro incluyen entumecimiento en la cara, brazo o pierna, confusión o dificultad para hablar, mareos inexplicables, visión borrosa, pérdida del equilibrio, dificultad para tragar, dolores de cabeza o incluso inconsciencia (similar a muchos de los síntomas descritos en compresión del tronco encefálico). Un enfoque lógico de esta preocupación podría incluir prescribir una aspirina infantil para bebés como terapia preventiva (como se hace en pacientes que tienen riesgo de accidentes cerebrovasculares tromboembólicos por otras razones), sin embargo, no hay información disponible que sugiera si esto debe hacerse en una base de rutina para personas con M-CM.
Manos y pies
Las correas o la fusión de la piel entre los dedos de los pies son muy comunes en M-CM, particularmente entre el segundo y tercer dedos. Esto se llama sindactilia. La sindactilia de los dedos es menos común y, cuando está presente, por lo general implica la fusión entre el tercer y cuarto dedo.
Además, M-CM puede causar polidactilia que generalmente es postaxial. La polidactilia y la sindactilia pueden ocurrir unilateral o bilateralmente y, si son graves, pueden corregirse fácilmente y no causar problemas de salud adicionales.
Comprar zapatos puede ser muy difícil para los niños con M-CM. Muchos tienen pies anchos e hinchados que a menudo son grandes y asimétricos. Algunas familias tienen que comprar zapatos de diferentes tamaños y, a veces, se deben hacer zapatos personalizados.
Los niños con M-CM también suelen tener uñas muy pequeñas y profundas (especialmente las uñas de los dedos del pie), que son difíciles de cortar y cuidar. Es posible que se necesite un podólogo ayudar en el cuidado y la supervisión adecuados
Tejido conectivo
Los niños con M-CM pueden tener anomalías en el tejido conectivo que afectan la piel y las articulaciones. El tejido conectivo es un material de tipo andamiaje que se une y sostiene el cuerpo. Ayuda a formar los tendones, las articulaciones, los ligamentos, los huesos y el cartílago. Parece haber un defecto en el tejido conectivo en M-CM. La piel a menudo se siente suave, pastosa y puede aparecer suelta, como si fuera demasiado grande para el cuerpo. Las articulaciones pueden ser levemente flojas o laxas. Las luxaciones conjuntas se han descrito al nacer. Los aspectos del tejido conectivo de M-CM son poco conocidos y se aclarará con el tiempo.
Rasgos faciales
Se han descrito varias características faciales únicas en individuos con M-CM. Aunque es difícil articular las similitudes sutiles entre las personas afectadas, al mirar las fotos, es claro que los niños con M-CM se parecen más entre sí que los miembros de sus propias familias. Las características faciales de M-CM no son necesariamente específicas del diagnóstico, aunque un médico con experiencia en el diagnóstico puede reconocer un patrón específico de las características faciales en M-CM más fácilmente que otro.
Si bien las características faciales individuales pueden ser muy variables, la prominencia frontal parece ser el hallazgo más frecuente y constante entre las personas afectadas. Otras características comúnmente reportadas incluyen:
- Asimetría facial
- Malformaciones capilares faciales en la línea media (vea la sección de la piel)
- Labios llenos
- Labio inferior evertido
- Mejillas llenas
- Orejas bajas
- Encías gruesas
- Boca grande
- Puente nasal bajo o deprimido
- Anteversión de las fosas nasales
- Hélices superiores superpuestas (el pliegue externo de la oreja)
- Nariz gruesa y carnosa
- Orejas grandes y carnosas o lóbulos de las orejas
- Philtrum largo
- Ojos hundidos
- Hipertelorismo (ojos muy separados)
Ortopédico
Los problemas ortopédicos en M-CM están relacionados con la asimetría de las extremidades y las articulaciones sueltas. Algunos pueden beneficiarse de la fisioterapia o incluso de los aparatos ortopédicos para la pierna o el tobillo si hay debilidad o flojedad importantes en los tobillos. Un levantamiento de zapatos también puede ayudar a aliviar problemas si una pierna es más larga que la otra. En algunos casos, se ha realizado un procedimiento quirúrgico llamado epifisialisis para evitar que las placas de crecimiento crezcan en la pierna más grande. Las anomalías del tejido conectivo en M-CM probablemente contribuyen a un mayor riesgo de luxaciones de cadera al nacer. La intervención quirúrgica a veces es necesaria. Si bien muchos de estos problemas ortopédicos y del tejido conectivo pueden contribuir a retrasos en el desarrollo, no parece que haya daños a largo plazo en las articulaciones, aunque la información disponible sobre esto es muy limitada.
Corazón
Un pequeño porcentaje de niños con M-CM están en riesgo de disrritmia y posible muerte súbita. La causa de este riesgo no se comprende. No está claro si esto puede deberse a la compresión del tronco encefálico debido al sobrecrecimiento del cerebro (ver sección del cerebro) que afecta el nervio vago o el tejido cardíaco anormalmente formado. Este riesgo parece ser mayor en los niños más pequeños con M-CM y parece haber menos riesgo a medida que maduran. Además, parece haber un mayor riesgo de defectos cardíacos congénitos. Algunas anormalidades informadas han incluido defectos septales, tetralogía de Fallot y coartación de la aorta. La incidencia de defectos cardíacos estructurales en M-CM no se conoce, pero parece ser relativamente poco común, tal vez se observa en el 5-10% de las personas afectadas. Debido a que la naturaleza exacta de los problemas cardíacos en M-CM sigue sin estar clara, se puede considerar una consulta con un cardiólogo pediátrico junto con un electrocardiograma (abreviado EKG). Un médico también puede considerar realizar un ecocardiograma específicamente para buscar defectos estructurales, especialmente si hay un soplo presente.
Pulmones
Si bien los pulmones parecen estar sanos en M-CM, puede haber problemas obstructivos de las vías respiratorias en este diagnóstico. El colapso de las vías respiratorias y / o de la caja de resonancia debido a un soporte inadecuado del cartílago de soporte dentro de la vía aérea se denomina traqueomalacia y laringomalacia, respectivamente. Estos problemas provocan un colapso parcial de las vías respiratorias durante la respiración o cambios posturales que pueden causar un bloqueo de las vías respiratorias.
Esto puede sonar como sibilancia o estridor durante la respiración normal o el sueño. Por qué esto ocurre en M-CM no está claro. Puede deberse a la hipotonía muscular, los problemas del tejido conectivo de la enfermedad o una combinación de factores. Estos problemas a menudo se ven también en niños no afectados. En niños “normales”, generalmente el cartílago de soporte en las vías respiratorias crece con la edad y los problemas obstructivos se resuelven por sí solos con el tiempo. Sin embargo, la historia natural de los problemas pulmonares en M-CM no está bien caracterizada.
Para diagnosticar un problema con la vía aérea, un especialista pulmonar puede optar por hacer un alcance para visualizar la estructura o función dinámica de la vía aérea. Para un niño con problemas respiratorios o pulmonares, también se puede ordenar una radiografía de tórax. Es importante estar al tanto de posibles problemas respiratorios subyacentes antes de cualquier intervención quirúrgica para prevenir posibles complicaciones durante la sedación. De lo contrario, no parece haber otras preocupaciones pulmonares importantes que no sean la posibilidad de apnea en el caso de la compresión del tronco del encéfalo (consulte la sección del cerebro).
Lo que se puede ver en el período prenatal y en el recién nacido
Se han observado signos de M-CM durante el embarazo. En la ecografía prenatal, se ha encontrado que algunos individuos diagnosticados posteriormente con M-CM tienen movimientos fetales disminuidos, grandes para la edad gestacional (LGA), hidrocefalia, protuberancia frontal, dedos de manos y pies adicionales, exceso de líquido amniótico, brazos o piernas asimétricos, derrame pleural e hidrops. Los niños afectados suelen ser grandes al nacer, incluida una cabeza grande. En raras ocasiones, los niños con M-CM nacen con un tamaño de cabeza normal, pero luego desarrollan macrocefalia. La hipoglucemia neonatal, las dificultades de alimentación, el retraso del crecimiento y la falta de crecimiento pueden manifestarse en el período neonatal además de las complicaciones respiratorias secundarias a la obstrucción de las vías respiratorias superiores (consulte la sección de los pulmones). No está claro por qué la hipoglucemia puede ocurrir en M-CM. La hipotonía también es una característica común que se presenta en el período del recién nacido y puede explicar la disminución de la actividad fetal.
Genética
Un artículo de revista de junio de 2012 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3408813/) informa que M-CM es causada por una mutación en un gen llamado PIK3CA. Esta mutación se encontró en 23 individuos con M-CM. En la mayoría de los casos, la mutación fue el resultado del mosaicismo somático. Esto significa que la mutación ocurrió después de que la división celular había comenzado y no fue heredada. En dos casos, se pensó que las mutaciones eran mutaciones en la línea germinal. No hay casos reportados de M-CM corriendo en familias. No hay casos reportados de individuos con M-CM que se hayan reproducido, por lo que se desconoce si M-CM podría ser heredado bajo esta circunstancia.
Las mutaciones PIK3CA estuvieron recientemente implicadas en otro síndrome de sobrecrecimiento llamado CLOVES, donde la mutación también se encuentra en un estado de mosaico. Existe una superposición de síntomas entre los dos síndromes, pero también tienen diferencias suficientes para distinguirlos entre sí en la mayoría de los casos. El significado completo de esta relación no se comprende en este momento.
PIK3CA también se encuentra mutado en cánceres humanos. Previo a este hallazgo genético, la necesidad de detectar cáncer de riñón (tumor de Wilms) y hepatoblastoma (cáncer de hígado) fue controvertida en M-CM. Los investigadores que publicaron este nuevo hallazgo genético ahora están recomendando la detección del tumor de Wilms para pacientes con M-CM, así como un mayor conocimiento de un posible riesgo elevado de cáncer para los médicos que atienden a pacientes con M-CM.
¿Cómo se diagnostica M-CM?
Si bien recientemente se identificó un marcador genético para M-CM, el diagnóstico generalmente se puede hacer basándose en las características observadas en un individuo. Haga clic aquí para obtener información sobre pruebas genéticas. Se han propuesto cuatro criterios diagnósticos diferentes para diagnosticar M-CM y se describen a continuación. Los dos primeros criterios se desarrollaron antes del cambio de nombre a M-CM y se refieren a cutis marmorata o cutis marmorata telangiectásica congénita como características clave de la enfermedad. Incluso con el uso de criterios de diagnóstico clínico, se debe considerar el cuadro clínico completo de un individuo. Los proveedores con experiencia personal en la evaluación de aquellos con M-CM y que están familiarizados con la variabilidad del diagnóstico clínico están preparados para tener un mayor éxito en la realización de un diagnóstico preciso.
Robertson Criteria
El primer autor en proponer criterios de diagnóstico para M-CM fue Robertson et al. en 2000. Este criterio sugería que un diagnóstico de M-CM podría realizarse en un individuo si presentaba macrocefalia congénita Y CMTC con al menos CUATRO de las siguientes características:
- Hipotonía neonatal
- Retraso en el desarrollo
- Defectos del tejido conectivo (laxitud articular, piel suave)
- Prominencia frontal
- Nevus facial flammeus en la línea media (malformación capilar)
- Sindactilia del dedo del pie
- Sobrecrecimiento segmentario (hemihiperplasia)
- Hidrocefalia.
Franceschini Criteria
Poco después de que se publicaran los criterios de Robertson, Franceschini et al. informó dos casos de M-CM en los que los cambios vasculares característicos de la piel se desvanecieron con el tiempo. Este fue el primer trabajo que claramente hizo esta observación. Franceschini consideró que la presencia o ausencia de CMTC / malformación capilar no era necesariamente necesaria para hacer un diagnóstico confiable, ya que, en algunas personas afectadas, este hallazgo era dependiente de la edad y podría resolverse con el tiempo. Por lo tanto, los criterios de Franceeschini estipulaban que un diagnóstico de M-CM debe considerarse en un individuo que tiene macrocefalia Y al menos DOS de las siguientes características:
- Crecimiento excesivo
- CMTC
- Angiomata
- Polidactilia / sindactilia
- Asimetría facial y de las extremidades
Wright Criteria
El tercer criterio presentado por Wright et al. en 2009 presentó una modificación de los dos criterios antes mencionados y básicamente combinó aspectos de ambos en un nuevo método propuesto de diagnóstico de M-CM. Estos autores conservaron la combinación de macrocefalia Y malformaciones capilares que ocurren en cualquier parte del cuerpo como características obligatorias con la presencia requerida de un número específico de características menores similares a las propuestas por Robertson. Sin embargo, los criterios de Wright son menos restrictivos que los de Robertson y afirman que un individuo debe tener al menos DOS de los siguientes, además de las características obligatorias mencionadas anteriormente:
- Asimetría de crecimiento
- Retraso en el desarrollo
- Malformación capilar facial en la línea media
- Hipotonía neonatal
- Sindactilia / polidactilia
- Prominencia frontal
- Hiperlaxitud articular / piel hiperelástica Hidrocefalia
Criterios Martínez-Glez***
Un cuarto criterio diagnóstico fue propuesto en 2010 por Martinez-Glez et al. y sugiere la presencia de al menos 3 criterios principales y dos menores. Este criterio tiene en cuenta el hecho de que las anomalías en la neuroimagen son extremadamente comunes en M-CM y representan un patrón reconocible de anormalidades:
|
Mayor (al menos 3) Macrocefalia malformaciones capilares Sobrecrecimiento / asimetría Alteraciones de neuroimágenes Ventriculomegalia Cavum septum pellucidum o cavum septum vergae Prominencia frontal Hernia amigdalar cerebelosa Asimetría cerebral y / o cerebelosa
|
Menor (al menos 2) Retraso en el desarrollo Malformación capilar en la línea media Hipotonía neonatal Sindactilia / polidactilia Anomalías del tejido conectivo Prominencia frontal Hidrocefalia
|
A diferencia de los tres criterios propuestos anteriormente, el artículo publicado por Martínez-Glez et al. es único en el sentido de que los autores intentaron probar sus criterios frente a casos notificados previamente para demostrar su capacidad de hacer un diagnóstico clínico preciso de M-CM. Los autores pudieron diagnosticar el 94% de los 136 casos reportados previamente de M-CM usando sus criterios. Esto sugiere que, hasta la fecha, puede ser el mejor conjunto de criterios clínicos disponibles para ayudar en el diagnóstico de M-CM.
Condiciones con funciones superpuestas
La mayoría de las afecciones comúnmente diagnosticadas erróneamente en individuos con M-CM incluyen aquellas con sobrecrecimiento y malformaciones vasculares vistas en la piel. El síndrome de CLOVE (S) se denomina por un acrónimo que significa: C (congénito) L (lipomatoso) O (sobrecrecimiento) V (malformaciones vasculares) E (nevo epidérmico) S (escoliosis y otra afectación esquelética). Esta condición se describió por primera vez en 2007 en un grupo de personas que habían sido diagnosticadas previamente con síndrome de Proteus. El CLAVO (S) puede causar malformaciones capilares, lipomas y hemihipertrofia, aunque por lo general no se asocia con hallazgos neurológicos significativos. CLOVES es causado por una mutación de mosaico en PIK3CA. Este es el mismo gen que está mutado en M-CM.
Cutis Marmorata Telangiectásica congénita (CMTC) se caracteriza por el marcado generalizado o segmentario de la piel debido a vasos sanguíneos prominentes. Parece cutis marmorata pero es mucho más pronunciada. Otras malformaciones vasculares como hemangiomas, parches de salmón y venas varicosas también pueden ocurrir en pacientes con CMTC. La causa es desconocida y aunque a menudo es un hallazgo aislado, existen riesgos de otras anomalías asociadas, como glaucoma y asimetría de las extremidades (con mayor frecuencia hemihipoplasia o sotobosque de la extremidad afectada). En general, CMTC aislado se ve muy diferente de M-CM. Debido a esto, no está claro si los trastornos comparten algo más que un parecido superficial. Las complicaciones neurológicas tales como macrocefalia, sobrecrecimiento cerebeloso, hidrocefalia y retraso en el desarrollo son menos comunes en comparación con M-CM.
El síndrome de Klippel-Trenaunay (KTS) se caracteriza por una tríada de hallazgos que incluyen malformaciones vasculares de flujo lento que involucran venas y capilares, un sistema linfático anormalmente desarrollado y hemihiperplasia. Típicamente, las extremidades y / o el tronco se ven afectados de manera aislada. El crecimiento excesivo de una parte del cuerpo afectada está presente en el nacimiento y a menudo es grave. Las manchas de vino de Oporto y las venas varicosas son comunes. De todos los diagnósticos enumerados aquí, es probable que KTS sea el que más diagnostique a las personas con M-CM. Generalmente, las personas con KTS no tienen macrocefalia y no tienen un retraso significativo en el desarrollo o problemas neurológicos. La causa es actualmente desconocida.
El síndrome de Parkes-Weber (SPW) es similar al síndrome de Klippel-Trenaunay. Sin embargo, en el SPW, las anomalías vasculares son lesiones de flujo rápido que implican conexiones anormales entre las arterias y las venas (malformaciones arteriovenosas o MAV). Se ha encontrado que algunos pacientes con SPW tienen mutaciones en un gen llamado RASA1.
El síndrome de Sturge-Weber (SWS) se caracteriza por malformaciones capilares de las meninges que generalmente se acompaña de manchas características en el vino facial (principalmente en el párpado superior y la frente) que existen en un patrón de distribución específico relacionado con el nervio facial. El trastorno a menudo tiene problemas neurológicos o convulsiones debido a la participación en el cerebro. En ocasiones puede haber malformaciones capilares de la piel debajo de la cabeza y el cuello. La causa es desconocida.
El síndrome de Beckwith-Wiedemann (BWS) es un trastorno de sobrecrecimiento relativamente común que incluye las siguientes características frecuentes: boca grande y lengua, onfalocele, hernia umbilical, pliegues o fosas oculares, hipoglucemia neonatal, malformaciones capilares y hemihiperplasia. Las pruebas genéticas pueden identificar la mayoría de los casos. Las personas con BWS generalmente no tienen las complicaciones neurológicas severas a menos que experimenten hipoglucemia severa en el período del recién nacido. BWS no tiende a causar las anormalidades vasculares generalizadas de la piel que se ven en M-CM. Del mismo modo, los pacientes con M-CM generalmente no tienen un mayor riesgo de onfalocele.
El síndrome de Proteus (PS) es un trastorno raro y complejo que involucra un crecimiento excesivo de partes del cuerpo junto con anomalías de los vasos sanguíneos y del sistema linfático, áreas localizadas de sobrecrecimiento óseo (hiperostosis), nevus epidérmicos, nevus del tejido conectivo y otros componentes del tejido conectivo como los músculos y la grasa. Los pacientes pueden tener tumores grasos benignos (lipomas) o falta de grasa debajo de la piel en ciertas partes del cuerpo. Esta es una condición rara y progresiva donde los niños generalmente nacen sin deformidades obvias y los adquieren con el tiempo. Esto distingue a PS de muchos otros síndromes de sobrecrecimiento, incluido M-CM.
Los criterios formales de diagnóstico para PS existen. Sin embargo, debido a que la enfermedad es poco conocida y rara, muchos médicos, incluso los genetistas experimentados, pueden tener dificultades para hacer el diagnóstico con precisión. Recientemente, una mutación en el gen AKT1 se ha asociado con PS. Esta mutación se observó en algunos, pero no en todos los tejidos de las personas afectadas, lo que confirma que esta condición se debe al mosaicismo genético. Se ha sugerido en el pasado que algunos individuos con PS tienen mutaciones en un gen llamado PTEN. Sin embargo, aquellos que claramente tienen PS cuando se aplica estrictamente el criterio de diagnóstico no tienen un defecto identificable en el gen PTEN. Este descubrimiento reciente aclara las diferencias moleculares entre PS debido al gen AKT1 y síndromes de tipo Proteus debido a mutaciones en PTEN.
Este nuevo descubrimiento de la asociación del gen AKT1 y PS debería disipar aún más cualquier posible vínculo directo entre las mutaciones PTEN y PS.
Hemihiperplasia: el síndrome de lipomatosis múltiple (HHML) se describió por primera vez en 1998 en un grupo de personas con asimetría corporal moderada y lesiones similares a las del lipoma debajo de la piel. Estas personas no cumplían los criterios de diagnóstico clínico para el síndrome de Proteus a pesar de muchas características superpuestas.
Síndromes Hamartoma relacionados con PTEN
PTEN es un gen que, cuando se altera, puede causar diferentes trastornos que se caracterizan por el desarrollo de crecimientos no cancerosos similares a tumores llamados hamartomas. En conjunto, los trastornos causados por PTEN se denominan síndromes de hamartoma relacionados con PTEN y se resumen a continuación. Algunos, pero no todos, los casos de las siguientes afecciones se deben a cambios identificables en el gen PTEN.
- El síndrome de Bannayan-Riley-Ruvalcaba (BRRS) se caracteriza por macrocefalia, hamartomas en los intestinos (poliposis hamartomatosa intestinal), lipomas y manchas pigmentadas en la piel del pene en los hombres. (imagen) (más información)
- El síndrome de Cowden es un trastorno poco común que causa hamartomas múltiples y un mayor riesgo de ciertos cánceres de tiroides, endometrio (revestimiento del útero) y mama. Los crecimientos hamartomatosos también pueden ocurrir en la piel, el tracto intestinal y las membranas mucosas (como el revestimiento de la boca y la nariz). Las personas afectadas a menudo tienen macrocefalia.
Síndrome similar a Proteus (PLS). Se ha encontrado que las personas con PLS tienen mutaciones en PTEN y se parecen clínicamente al síndrome de Proteus. Este es un grupo variable de entidades clínicas y el diagnóstico de un individuo puede ser difícil a menos que el proveedor tenga experiencia con la enfermedad.
El síndrome de MPPH (megalencefalia-polimicrogiria-polidactilia-hidrocefalia) es una enfermedad que se describió por primera vez en 2004. El MPPH comparte algunas características cerebrales M-CM que incluyen megalencefalia, polimicrogiria e hidrocefalia, como su nombre lo indica. Carece de anomalías vasculares y hemihiperplasia. El gen mutado implicado en MPPH ha sido identificado como PIK3R2, que está estrechamente relacionado con el gen mutado en M-CM, PIK3CA. El descubrimiento de genes para MPPH se publicó en el mismo documento que el de M-CM.
El síndrome de Costello se caracteriza por retraso en el desarrollo y discapacidad intelectual, rasgos faciales toscos distintivos, cabello fino rizado o escaso, pliegues sueltos de piel adicional (especialmente en las manos y los pies), hipotonía y articulaciones inusualmente flexibles. Las anomalías cardíacas son comunes, incluida la taquicardia, los defectos cardíacos estructurales y la miocardiopatía hipertrófica. Los bebés con síndrome de Costello tienden a ser más grandes que el promedio al nacer, incluida la macrocefalia con protuberancia frontal progresiva. No tienen los mismos cambios en la piel vascular que se observan en M-CM ni las malformaciones características de los dígitos.
Existe una superposición considerable con M-CM en el síndrome de Costello en términos de hallazgos neurológicos en los estudios de imágenes cerebrales. Los hallazgos neurológicos comunes en Costello incluyen malformaciones de Chiari I, ventriculomegalia / hidrocefalia evolutiva y progresiva, abducción de la fosa posterior y hernia de las amígdalas cerebelosas. El síndrome de Costello es causado por mutaciones en el gen HRAS y hay pruebas clínicas disponibles. El trastorno es lo suficientemente diferente de M-CM que un genetista clínico experimentado debería ser capaz de diferenciar las condiciones.
El síndrome de Sotos es un trastorno de crecimiento excesivo con características que incluyen macrocefalia, rasgos faciales característicos, sobrecrecimiento del cuerpo, problemas de aprendizaje, problemas de conducta, convulsiones, escoliosis, defectos congénitos del corazón y del riñón. La asimetría o hemihiperplasia, la sindactilia / polidactilia y las anormalidades de la piel vascular no son características comunes en este síndrome. El síndrome de Sotos se debe a defectos en el gen NSD1 y las pruebas genéticas están disponibles.
El síndrome de Simpson-Golabi-Behmel (SGBS) es una afección genética asociada con macrosomía, características faciales distintivas, macrocefalia, defectos cardíacos, anomalías gastrointestinales y genitourinarias, hallazgos musculoesqueléticos que incluyen la luxación congénita de la cadera y anomalías digitales que incluyen polidactilia y sindactilia. Las personas afectadas tienen una afectación neurológica variable, que va desde un retraso mental normal a severo con o sin anomalías estructurales del cerebro. SGBS se debe a cambios en el gen GPC3 y las pruebas genéticas están disponibles. A diferencia de los otros trastornos en esta lista, SGBS afecta principalmente a los niños debido al hecho de que es una condición ligada a X. Esto significa que el gen GPC3 se encuentra en el cromosoma X.
El síndrome de MOMO es una condición de sobrecrecimiento extremadamente rara y se han reportado muy pocos casos. El nombre es un acrónimo de los cuatro aspectos principales del trastorno: macrosomía, obesidad, macrocefalia y anomalías de los ojos. La causa es desconocida